How the GRC should address rare bases, as well as common
bases that result in non-functional alleles (a.k.a. polymorphic pseudogenes), in
the reference assembly is a substantially more complex task than dealing with erroneous bases. This is made even
more so by the wide range of opinions held by assembly users, which include:
- Most common allele
- Ancestral allele
- Coding allele
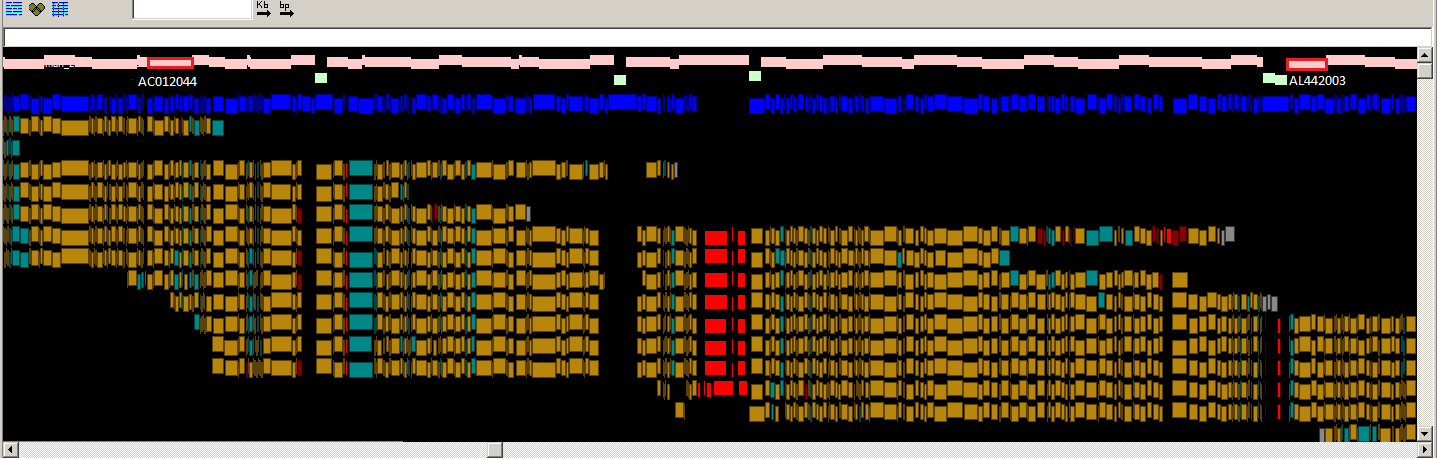
The GRC currently favors a model in which haplotypic integrity is retained within blocks of linkage disequilibrium (LD) as best possible, every base is found at an MAF >5% in some population (i.e. no universally rare alleles) and coding alleles are favored over non-coding alleles, so long as they too are not universally rare. However, additional analyses will be performed before any bases changes are made. Examples of genomic regions where the existing reference base is associated with disease (ASPN, PMID:15640800) or non-coding variants (CYP3A5, PMID:11279519) are presented below in Figures 1 and 2. In the former case, the reference base is the minor allele, while in the latter, it is the major allele. We invite you to consider examples such as these as you form your own views of what should be represented in the reference assembly. If you have questions or concerns about base updates for GRCh38, let us know!
Fig.1

Fig. 2

Fig. 2. Zoomed-in graphical view of the CYP3A5 gene in GRCh37. The
assembly sequence is shown at top. The CYP3A5 gene is shown in green. The
highlighted base in the GRCh37 reference assembly represents the major allele
in many populations. This allele creates cryptic splice site that disrupts the
reading frame of CYP3A5 and results in a non-coding transcript. However, in
other populations, the coding allele is the major allele.